It's been interesting.
Here is the code and some nice output.
#load flowCore which allows the import of FCS files.
source("http://bioconductor.org/biocLite.R")
biocLite("flowCore")
library(flowCore)
# import the data
ntl<-read.FCS("A01 NTL.fcs", alter.names = TRUE)
n <- exprs(ntl) # converts the data into a matrix
cd40L<-read.FCS("A02 CD40L.fcs", alter.names = TRUE)
c <- exprs(cd40L) # converts the data into a matrix
#BASE GRAPHICS
# Use base graphics of R to display FSC/SSC dot plots.
par(mfrow=c(1,2)) #allows to plots to be placed side to side
plot(n[,1], n[,2],
pch = ".",
ylim=c(0,1000000),
xlim=c(0,10000000),
xlab="FSC-A",
ylab="SSC-A",
main="NTL") # this works
plot(c[,1], c[,2],
pch = ".",
ylim=c(0,1000000),
xlim=c(0,10000000),
xlab="FSC-A",
ylab="SSC-A",
main="CD40L") # this works
This is the plot produced:

#GGPLOT2
#Can also use ggplot2 to draw dot plots
library(ggplot2)
#seems ggplot only likes data frames
ndf <- as.data.frame(n)
cdf <- as.data.frame(c)
#make the plotting objects using ggplot
p1<- ggplot(ndf, aes(x=FSC.A, y=SSC.A)) +
geom_point(shape=".", colour="red") +
ylim(0, 500000) + xlim(0,5000000) +
ggtitle("Samp 001, NTL & IL4")
p2<- ggplot(cdf, aes(x=FSC.A, y=SSC.A)) +
geom_point(shape=".", colour="red") +
ylim(0, 500000) + xlim(0,5000000) +
ggtitle("Samp 001, CD40L & IL4")
multiplot(p1,p2, cols=2)
This is the output

# 2D density plots
#with contour lines
p1 <- ggplot(ndf, aes(x=FSC.A, y=SSC.A)) +
geom_point(shape=".", colour="red") +
ylim(0, 500000) + xlim(0,5000000) +
ggtitle("Samp 001, NTL & IL4") +
geom_density2d(colour="black", bins=5)
p2 <- ggplot(cdf, aes(x=FSC.A, y=SSC.A)) +
geom_point(shape=".", colour="red") +
ylim(0, 500000) + xlim(0,5000000) +
ggtitle("Samp 001, NTL & IL4") +
geom_density2d(colour="black", bins=5)
multiplot(p1,p2, cols=2)

#with colour indicating density
ggplot(ndf, aes(x=FSC.A, y=SSC.A)) +
geom_point(shape=".", colour="red") +
ylim(0, 500000) + xlim(0,5000000) +
ggtitle("Samp 001, NTL & IL4") +
stat_density2d(aes(alpha=..density..), geom="raster", contour = FALSE)

Some very useful help from here:
http://www.talkstats.com/showthread.php/52675-ggplot2-contour-chart-plotting-concentrations/page2
#with more colours indicating density
colfunc <- colorRampPalette(c("white", "lightblue", "green", "yellow", "red"))
ggplot(ndf, aes(x=FSC.A, y=SSC.A)) +
ylim(0, 500000) + xlim(0,5000000) +
stat_density2d(geom="tile", aes(fill = ..density..), contour = FALSE) +
scale_fill_gradientn(colours=colfunc(400)) +
geom_density2d(colour="black", bins=5)
Cells on NTL with nice colours
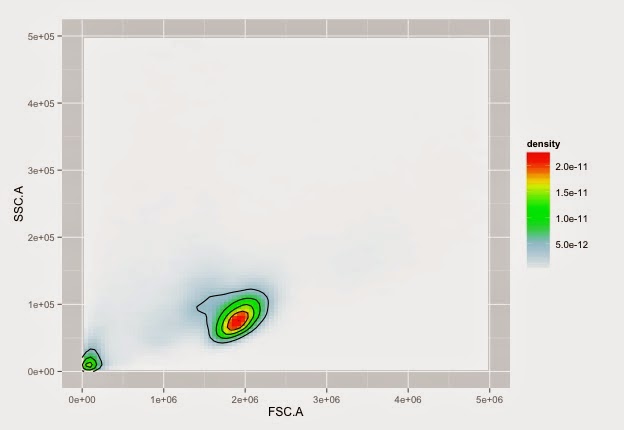
ggplot(cdf, aes(x=FSC.A, y=SSC.A)) +
ylim(0, 500000) + xlim(0,5000000) +
stat_density2d(geom="tile", aes(fill = ..density..), contour = FALSE) +
scale_fill_gradientn(colours=colfunc(400)) +
geom_density2d(colour="black", bins=5)
Cells on CD40L with nice colours

#function to allow more than one ggplot on a page
multiplot <- function(..., plotlist=NULL, file, cols=1, layout=NULL) {
library(grid)
# Make a list from the ... arguments and plotlist
plots <- c(list(...), plotlist)
numPlots = length(plots)
# If layout is NULL, then use 'cols' to determine layout
if (is.null(layout)) {
# Make the panel
# ncol: Number of columns of plots
# nrow: Number of rows needed, calculated from # of cols
layout <- matrix(seq(1, cols * ceiling(numPlots/cols)),
ncol = cols, nrow = ceiling(numPlots/cols))
}
if (numPlots==1) {
print(plots[[1]])
} else {
# Set up the page
grid.newpage()
pushViewport(viewport(layout = grid.layout(nrow(layout), ncol(layout))))
# Make each plot, in the correct location
for (i in 1:numPlots) {
# Get the i,j matrix positions of the regions that contain this subplot
matchidx <- as.data.frame(which(layout == i, arr.ind = TRUE))
print(plots[[i]], vp = viewport(layout.pos.row = matchidx$row,
layout.pos.col = matchidx$col))
}
}
}
